3 Methoden
3.1 DNA-Isolierung3.2 Reinigen und Fällung von DNA
3.3 Absorptionsmessungen
3.4 Enzymatische DNA-Behandlung
3.5 Transformation
3.6 a-Komplementation
3.7 Agarose-Gelelektrophorese
3.8 Elution von DNA aus Agarosegel
3.9 Radioaktive DNA-Sequenzierung mit 35S
3.10 Nachweis von homologen DNA-Sequenzen
3.1.1 Isolierung von Gesamt-DNA
Zur Isolierung von Gesamt-DNA wurde die von Ausubel beschriebene Methode verwendet [Ausubel et al., 1992].
1.5 ml einer ÜK wurden 5 min in einer HETTICH-Kühlzentrifuge bei 4°C und 12 000 rpm (wie auch bei allen folgenden Zentrifugationsschritten) abzentrifugiert. Das Pellet wurde in 567 µl TE-Puffer resuspendiert und die entstandene Zellsuspension mit 30 µl 10 % (w/v) SDS und 3 µl Proteinase K (20 mg/ml) versetzt. Durch mehrmaliges Kippen wurde der Ansatz durchmischt und bis zur Klärung des Lysates für ca. 1 h bei 37°C inkubiert. Die hochviskose Suspension wurde durch fünfmaliges Aufziehen mit einer 1 ml-Einwegspritze (0.9 mm-Kanüle) homogenisiert und mit 100 µl zugesetzter 5 M NaCl-Lösung gut durchmischt. Nach Zugabe von 80 µl CTAB/NaCl, gründlicher Durchmischung und Inkubation für 10 min bei 65°C, wurden 0.7 ml Chloroform-Isoamylalkohol zugegeben, kurz gevortext und für 5 min zentrifugiert. Zur Entfernung von Proteinen wurde die Oberphase in ein neues Eppendorfgefäß überführt und eine Phenolextraktion durchgeführt. Es schloß sich eine Fällung der DNA durch Zugabe des 0.6fachen Volumens Isopropanol an. Nach 15minütiger Zentrifugation wurde das erhaltene DNA-Pellet mit 70 % EtOH gewaschen, in der Speed-Vac getrocknet und in 40 µl TE-Puffer für 10 min bei 37°C resuspendiert. Von dieser Lösung waren 2-5 µl für eine analytische Gelelektrophorese ausreichend.
3.1.2 Plasmidisolierung im kleinen Maßstab
3.1.2.1 Kurzlysat mittels alkalischer Lyse
Für die schnelle Gewinnung von Plasmid-DNA zur Klonierung und Sequenzierung wurde die Methode von Birboim, beschrieben bei Sambrook, in modifizierter Form angewandt [Birnboim u. Doly, 1979; Sambrook et al., 1989].
In einem Eppendorfgefäß wurden 1.5 ml einer über Nacht gewachsenen Bakterienkultur in einer HETTICH-Kühlzentrifuge bei 4°C und 12 000 rpm (wie auch bei allen weiteren Zentrifugationsschritten) kurz abzentrifugiert und der Überstand mittels einer Wasserstrahlpumpe abgesaugt. Das so getrocknete Pellet wurde in 100 µl GET-Puffer resuspendiert. Nach fünfminütiger Inkubation bei RT wurden 200 µl frisch zubereiteter Lysismix zugegeben, um die Zellen zu lysieren. Der Inhalt des Gefäßes wurde durch mehrmaliges Kippen gemischt und 5 min auf Eis inkubiert. Nach Zugabe von 150 µl K-Acetatlösung wurde erneut gemischt und weitere 5 min auf Eis inkubiert. Der Niederschlag wurde 10 min abzentrifugiert, der DNA-haltige Überstand durch eine Phenolextraktion von Proteinresten gereinigt und nach anschließender EtOH-Fällung wurde das getrocknete DNA-Pellet in 40 µl TER-Puffer gelöst. Von der erhaltenen DNA-Lösung waren 2-10 µl für eine analytische Gelelektrophorese ausreichend.
3.1.2.2 Kurzlysat mittels 'LiCl-Single-Step' Methode
Die 'LiCl-Single-Step' Methode zur Isolierung von Plasmid-DNA von He stellt aufgrund des geringeren Zeit- und Materialaufwandes eine effektive Alternative zur Methode nach Birnboim und Doly dar und wurde für das 'Schnell-Screening' zur Plasmidisolierung aus einer Vielzahl von Klonen verwendet [He et al., 1989].
1.5 ml einer ÜK wurden in einer HETTICH-Kühlzentrifuge bei 12 000 rpm (wie auch auch bei allen folgenden Zentrifugationsschritten) abzentrifugiert. Das Bakterienpellet wurde in 100 µl TELT-Puffer resuspendiert und mit 1 Vol. Phenol-Chloroform versetzt. Der Ansatz wurde 15 sec kräftig geschüttelt und 1 min bei 22°C zentrifugiert. Die wäßrige Oberphase wurde abgenommen und die darin enthaltene DNA durch Zugabe von 2 Vol. 96 % EtOH ausgefällt. Nach zehnminütiger Zentrifugation wurde das DNA-Pellet mit 1 ml 96 % EtOH gewaschen, kurz zentrifugiert, in der Speed-Vac für 10 min getrocknet und in 30 µl TER-Puffer gelöst. Von der erhaltenen DNA-Lösung waren 2-8 µl für eine analytische Gelelektrophorese ausreichend.
3.1.3 Plasmidisolierung im großen Maßstab
3.1.3.1 Alkalische Lyse nach Birnboim und Doly
Diese Methode zur Plasmidisolierung wurde in modifizierter Form für Plasmide kleiner als 100 kb durchgeführt [Birnboim und Doly, 1979].
200 ml einer über Nacht gewachsenen Bakterienkultur wurden in JA14-Bechern in einer BECKMANN J21C-Zentrifuge bei 4°C und 8 000 rpm (wie auch bei den nächsten Zentrifugationsschritten) 10 min abzentrifugiert. Das Zellpellet wurde mit 50 ml 25 mM Tris-HCl-Lösung (pH 8.0) gewaschen und nach erneuter Zentrifugation in 5 ml 50 mM Tris-HCl-Lösung (pH 8.0) resuspendiert. Nach Zugabe von 5 ml GETL-Puffer erfolgte eine Inkubation von 30 min auf Eis. Anschließend wurden 20 ml frisch angesetzter Lysismix zugegeben und nach Durchmischung erfolgte eine Inkubation von mindestens 15 min auf Eis. Dem Ansatz wurden daraufhin 15 ml K-Acetatlösung zugesetzt und nach 60minütiger Inkubation auf Eis wurde die ausgefällte Proteinfraktion durch einen 20minütigen Zentrifugationsschritt pelletiert. Der Überstand wurde sorgfältig in einen neuen JA14-Becher überführt und die darin enthaltene DNA durch Zusatz von 0.6 Vol. eiskaltem Isopropanol und 15minütige Inkubation bei -20°C gefällt. Nach 15minütiger Zentrifugation wurde der Überstand vorsichtig dekantiert und das getrocknete DNA-Pellet in 3 ml TEN-Puffer resuspendiert, in Corex-Röhrchen überführt, mit 4.5 g CsCl und 0.5 ml EtBr-Lösung (5 mg/ml) versetzt und mit TEN-Puffer zu einem Endvolumen von 4 ml aufgefüllt. Nach kräftigem Schütteln und vollständiger Lösung des Cäsiumchlorids wurde mit Corex-Einsätzen für den JA20 Rotor 20 min zentrifugiert. Nach Überführung des DNA-haltigen Überstandes in Quickseal-Röhrchen mußten diese vor dem Zuschweißen exakt austariert werden. Zur Einstellung des Ethidiumbromid-Cäsiumchlorid-Dichtegradienten wurde 18 h bei 15°C und 45 000 rpm mit einem vTi65-Rotor zentrifugiert (BECKMANN-L8-55-Ultrazentrifuge). Die Plasmidbande wurde unter UV-Licht mit Hilfe einer 1 ml-Einwegspritze abgezogen und zur Entfernung des Cäsiumchlorids für 2 h bei 4°C gegen TE-Puffer dialysiert. Durch Extraktion mit 1 Vol. Phenol-Tris und 15minütige Zentrifugation bei 4°C und 12 000 rpm wurde das verbliebene Ethidiumbromid entfernt (HETTICH-Kühlzentrifuge). Die DNA-haltige Oberphase wurde über Nacht gegen TE-Puffer dialysiert. Die Konzentration und Reinheit der DNA-Lösung wurde anschließend durch eine photometrische Messung und eine Analyse mittels Agarose-Gelelektrophorese kontrolliert.
3.1.3.2 Modifizierte Präparation nach Kado und Liu
Zur Isolierung von Plasmiden, deren Größe im Bereich von 100-200 kb lag, wurde eine modifizierte Methode nach Kado u. Liu angewandt [Kado u. Liu, 1981]. Dabei war besonders zu beachten, daß die DNA-haltige Flüssigphase zur Vermeidung von Scherkräften, die die Ausbeute an intakter Plasmid-DNA herabgesetzt hätten, stets sehr vorsichtig gehandhabt werden mußte.
50 ml einer ÜK wurden in JA-20 Zentrifugenröhrchen im JA-20 Rotor einer BECKMANN J21C-Zentrifuge (wie auch bei allen weiteren Zentrifugationsschritten) 10 min bei 6 000 rpm abzentrifugiert und in 8 ml TAE-Puffer resuspendiert. Zur Lyse der Zellen wurden 8 ml Lyse-Puffer hinzugegeben und die Suspension vorsichtig geschwenkt. Nach Durchmischung des Zell-Lysates wurde 1 h bei 65°C inkubiert. Anschließend wurden 2.5 ml auf 65°C vortemperierte 2.5 M NaCl-Lösung zugegeben. Erneut wurde die Lösung durch Schwenken gemischt und 20 min auf Eis abgekühlt. Nach dem Auffüllen der Zentrifugenröhrchen mit 1 Vol. saurem Phenol, Durchmischen und Zentrifugation für 15 min bei RT und 10 000 rpm wurde die wäßrige Oberphase mit einer unten erweiterten Pasteurpipette abgehoben und in einen neuen Zentrifugenbecher überführt. Die darin enthaltene Plasmid-DNA wurde einer EtOH-Fällung unterzogen und in 3 ml TEN-Puffer resuspendiert. Nach Ethidiumbromid-Cäsiumchlorid-Dichtegradientenzentrifugation, Phenolextraktion und Dialyse (wie unter 3.1.3. beschrieben) wurde die Konzentration und Reinheit der DNA-Lösung photometrisch und gelelektrophoretisch bestimmt.
3.2 Reinigen und Fällung von DNA
3.2.1 Proteinase K - Behandlung
Zur Inaktivierung und Entfernung von Phenol-stabilen Proteinen (wie dem Restriktions-enzym XhoI), die nicht durch eine Phenolextraktion entfernt werden können, wurde der DNA-Lösung Proteinase K (1 mg/ml) und SDS zu einer Endkonzentration von 1 % (w/v) zugesetzt. Nach mehrstündiger Inkubation der Lösung bei 60°C erfolgte eine Phenol-extraktion mit anschließender EtOH-Fällung.
3.2.2 Phenolextraktion
Zur Entfernung von Verunreinigungen aus DNA-Proben, z.B. Proteinen, wurden diese nacheinander mit je 1 Vol. Phenol-Tris und Phenol-Chloroform ausgeschüttelt und 2 min bei RT inkubiert. Nach zehnminütiger Zentrifugation (wie auch bei allen weiteren Zentrifugationsschritten bei 4°C und 12 000 rpm in einer HETTICH-Kühlzentrifuge durchgeführt) wurde der wäßrige Überstand in ein jeweils neues Reaktionsgefäß überführt. Dieser Schritt war so oft zu wiederholen, bis die Phenol/Wasser-Phasengrenze frei von weißen Proteinresten war. Um Phenol restlos aus der Probe zu entfernen, wurde die DNA-Lösung zweimal mit 0.5 Vol. Chloroform-Isoamylalkohol ausgeschüttelt, 1 min abzentrifugiert und die wäßrige Oberphase in ein neues Reaktionsgefäß überführt.
3.2.3 Dialyse
Um Verunreinigungen, wie z.B. Ethidiumbromid oder Cäsiumchlorid, aus einer DNA-Probe zu entfernen, wurde mindestens 1 h gegen 2 l TE-Puffer dialysiert. Die Dialyseschläuche wurden vor Gebrauch einmal in 5 % Na2CO3 und zweimal in TE-Puffer ausgekocht, in TE-Puffer autoklaviert und anschließend bei 4°C gelagert.
3.2.4 Ethanolfällung
Die Fällung der DNA aus wäßrigen Lösungen erfolgte durch Zugabe von 0.1 Vol. Na-Acetatlösung und 2 Vol. 96 % EtOH und anschließender Inkubation für 2 h bei -80°C oder über Nacht bei -20°C. Die gefällte DNA wurde durch Zentrifugation pelletiert, mit ca. 400µl 70 % EtOH gewaschen und nach Trocknung in der Speed-Vac in einem geeigneten Volumen TE-Puffer oder ddH2O gelöst.
Spektralphotometrische Messungen wurden mit einem LKB Ultrospec II Spektral-photometer durchgeführt.
DNA-Konzentrationen, bzw. der Reinheitsgrad der DNA wurde durch Messung der Absorption bei 260 und 280 nm in Quarzküvetten ermittelt. Eine Absorptionseinheit bei 260 nm entspricht bei einer Schichtdicke von 1.0 cm etwa 50 µg/ml doppelsträngiger DNA. Lag der Quotient der Absorptionsmessungen (A260/A280) nicht im Bereich von 1.6 bis 2.0, wurde die DNA durch Behandlung mit Proteinase K aufgereinigt.
Zur Bestimmung der Bakteriendichte durch Trübungsmessung wurde die optische Dichte von 1 ml der Zellsuspension bei einer Wellenlänge von 578 nm in Polystyrol-halbmikroküvetten bestimmt. Als Referenz diente im Einzelfall immer das unbeimpfte Medium. Überstieg die optische Dichte einen Meßwert von 0.5, so wurde die Zellsuspension verdünnt, da nur in einem OD-Bereich von 0.1 bis 0.5 eine lineare Beziehung zwischen Zelldichte und Trübung besteht.
3.4 Enzymatische DNA-Behandlung
3.4.1 Hydrolyse mit Restriktionsenzymen
Die Hydrolyse von DNA mit Typ II -Restriktionendonukleasen [Arber, 1978] erfolgte entsprechend den Empfehlungen des Herstellers.
Dabei wurde die zu schneidende DNA-Probe mit ddH2O und dem jeweiligen Restriktionspuffer in einem geeigneten Verhältnis, entsprechend der Pufferkonzentration, zu einem Endvolumen von 10, 20 oder 30 µl aufgefüllt. Je nach eingesetzter DNA-Menge wurden anschließend 1-5 Units des jeweiligen Restriktionsenzyms zugesetzt. Mit Ausnahme der Restriktionsverdaus mit SmaI, die bei 30°C inkubiert wurden, erfolgte bei allen anderen verwendeten Restriktionsenzymen eine ein- bis zweistündige Inkubation bei 37°C. Für die Hydrolyse mit BamHI war zu beachten, daß nach mehr als einstündiger Inkubation und bei hoher Glycerinkonzentration Sternaktivität des Enzyms auftreten kann; entsprechend wurden diese Restriktionsverdaus für eine Stunde in einem erhöhten Volumen und bei erhöhter Enzymkonzentration durchgeführt. Bei der Hydrolyse eines DNA-Moleküls mit mehreren Restriktionsenzymen erfolgte jeweils zwischen den Einzelverdaus eine EtOH-Fällung.
3.4.2 Dephosphorylierung
Um bei Klonierungsreaktionen eine Rezirkularisierung des linearisierten Vektors zu verhindern, wurde der entsprechend linearisierte Vektor nach einer modifizierten Methode nach Sambrook mit alkalischer Shrimp-Phosphatase behandelt, wobei die für eine Eigenligation des Vektors essentiellen 5'-Phosphatgruppen entfernt wurden [Sambrook et al., 1989].
Konkret wurde die mit einem Restriktionsenzym hydrolysierte Vektor-DNA gefällt und in einer Konzentration von ca. 20 µg/ml in einfach konzentriertem Phosphatasepuffer (Fa. USB) aufgenommen. Je nach Beschaffenheit der DNA-Enden wurde alkalische Shrimp-Phosphatase zugegeben: Bei 3'-überstehend: 0.5 U; glatt: 0.2 U; 5'-überstehend: 0.1 U (Angaben jeweils für 1 pmol DNA-Enden). Nach einstündiger Inkubation bei 37°C erfolgte die Inaktivierung der alkalischen Shrimp-Phosphatase durch 15minütige Inkubation bei 65°C. Die dephosphorylierte Vektor-DNA konnte nach EtOH-Fällung direkt zur Ligation verwendet werden.
3.4.3 ExonukleaseIII - Behandlung
Die ExonukleaseIII von E.coli ermöglicht den unidirektionalen Abbau von DNA [Henikoff, 1984]. Dabei verkürzt sie doppelsträngige DNA mit stumpfen oder 5'-überstehenden Enden, nicht aber mit 3'-überstehenden Enden. Der Abbau erfolgt vom 3'-Ende in 5'-Richtung und durch Kombination zweier geeigneter Restriktionsenzyme ist es möglich, ein in die 'Polycloning site' eines Vektors kloniertes Insert zu verkleinern, während der Vektor vor einem Abbau geschützt wird.
Zur Herstellung und Isolierung von Deletionsklonen für die DNA-Sequenzierung wurde der Double-stranded Nested Deletion Kit von LKB/Pharmacia gemäß den Angaben des Herstellers verwendet.
Konkret wurden ca. 3 µg Plasmid-DNA mit zwei verschiedenen Restriktionsenzymen hydrolysiert, die auf der Vektorseite ein schützendes 3'-Ende und auf der Seite des zu deletierenden Inserts ein abbaubares 5'-Ende erzeugten. Nach Hitzeinaktivierung der Restriktionsendonukleasen, EtOH-Fällung und Resuspendierung in 20 µl ddH2O wurde die NaCl-Konzentration der DNA-Lösung durch Zugabe von 20 µl ExoIII-Puffer auf 75 mM eingestellt, was einen kontrollierbaren DNA-Abbau durch die ExonukleaseIII gewährleistet. Zur Erzeugung von Deletionen in 200-bp-Intervallen lag die Inkubationstemperatur der folgenden Behandlung mit ExonukleaseIII bei 25°C. Entsprechend wurde der Reaktionsansatz für 3 min auf 25°C vorinkubiert, ein 2 µl Aliquot als Nullwert mit 3 µl S1-Mix vermischt und auf Eis gestellt. Die Reaktion wurde durch Zugabe von 1 µl ExonukleaseIII gestartet. Im Abstand von 6 min wurden daraufhin insgesamt zehn Aliquots entnommen, mit je 1 µl S1-Mix versetzt und ebenfalls auf Eis gestellt. Nach Beendigung der einstündigen Versuchsreihe erfolgte eine 30minütige Inkubation sämtlicher Aliquots bei RT. Durch ExonukleaseIII-Behandlung einzelsträngige DNA-Bereiche wurden dabei durch die S1-Nuklease abgebaut. Das halbe Volumen (3 µl) des Reaktionsansatzes eines jeden Zeitwertes wurde dann auf einem Agarosegel aufgetrennt und analysiert, die andere Hälfte durch Zusatz von 17 µl Ligase-Mix und mindestens zwölfstündige Inkubation bei 16°C rezirkularisiert. Die Ergebnisse der Elektrophorese gaben einen Hinweis, welche Aliquots sich nach der Ligation aufgrund ihrer Deletionsgröße am besten für die Transformation eignen würden. Kompetente E.coliDH5a-Zellen wurden mit den entsprechenden Ligationsansätzen transformiert und auf Selektivmedien ausplattiert. Von den erhaltenen Transformanten wurde Plasmid-DNA isoliert und die Größe der Plasmide durch Restriktionsverdau überprüft.
3.4.4 Ligation
Zum Einbau von Insert-DNA in einen Vektor wurde die T4 DNA-Ligase der Fa. BRL verwendet. Dieses Enzym katalysiert die Bildung einer Phosphodiesterbindung zwischen den 3'-Hydroxy- und 5'-Phosphatenden zweier DNA-Fragmente.
Für die Reaktion wurden in einem Volumen von 20 µl 10-100 ng Vektor-DNA und ein zwei- bis dreifach molarer Überschuß der Insert-DNA (bezogen auf die Vektorkonzentration) mit 4 µl Ligasepuffer (Fa. BRL) und 0.1-0.2 Units T4 DNA-Ligase gemischt. Anschließend wurde für mindestens 12 h bei 16°C inkubiert. Zur Transformation des Ligationsansatzes wurde dieser 1:5 verdünnt, um störende Effekte durch eine zu hohe Glycerinkonzentration zu vermeiden.
Zur Transformation von E.coli-Zellen wurde die von Cohen entwickelte CaCl2-Methode in einer modifizierten Form angewandt [Cohen et al., 1972].
Zur Herstellung kompetenter Zellen wurden 50 ml LB-Medium mit 1 ml ÜK angeimpft, bei 30°C ca. 3 h bis zu einer OD550 von 0.65-0.75 inkubiert und in JA20-Röhrchen für den JA20 Rotor einer BECKMANN J21C-Zentrifuge bei 4°C und 5 000 rpm (wie auch bei den folgenden Zentrifugationsschritten) 10 min abzentrifugiert. Das Zellpellet wurde in 20 ml eiskalter CaCl2-Lösung (100 mM) resuspendiert und mind. 20 min auf Eis inkubiert. Nach zehnminütiger Zentrifugation wurden die pelletierten Zellen in 4 ml eiskalter CaCl2-Lösung (100 mM) resuspendiert. Um dauerkompetente Zellen herzustellen, wurde der Zell-suspension 1 ml steriles Glycerin zugegeben. Von diesem Gemisch wurden jeweils 100 µl in Eppendorfgefäße aliquotiert, in flüssigem Stickstoff eingefroren und bei -80°C aufbewahrt.
Vor der Transformation wurde ein Aliquot der dauerkompetenten Zellen 10 min im Eisbad aufgetaut und anschließend mit 20-200 ng zu transformierender DNA versetzt. Das Gemisch wurde mindestens 30 min auf Eis inkubiert, gefolgt von einem Hitzeschock bei 42°C für 90 sec. Nach zehnminütiger Inkubation auf Eis wurde 1 ml LB-Medium zugegeben und 1 h bei 37°C inkubiert. Die Zellen wurden daraufhin mit 1 ml LB-Medium gewaschen (Zentrifugation bei 4°C und 12 000 rpm in einer HETTICH-Kühlzentrifuge), in 20 µl Restvolumen resuspendiert und auf einer LB-Platte mit entsprechendem Antibiotikum als Selektionsmarker ausplattiert.
Die Screening-Methode der a-Komplementation wurde zur Kontrolle einer erfolgreichen Klonierung und Transformation verwendet und beruht auf dem qualitativen Nachweis von ß-Galactosidase-Aktivität [Miller, 1972]. Der Klonierungvektor pUC18 weist eine 'Polycloning-site' vor dem ß-gal-a-Fragment auf, während der Rezipientenstamm E.coliDH5a im lacZ teildeletiert ist und lediglich über das ß-gal-w-Fragment verfügt. Nach der Transformation von pUC18 wird das LacZ von E.coliDH5a durch die a-Kette des pUC18 komplementiert - auf IPTG/X-Gal-Platten sichtbar durch eine blaue Kolonie-färbung. Bei Insertion von Fremd-DNA in die 'Polycloning-site' und damit verbundener Zerstörung des lacZ von pUC18 zeigt sich auf IPTG/X-Gal-Platten eine weiße Färbung der Kolonien.
Auf eine LB-Platte mit einem geeignetem Antibiotikum als Selektionszusatz wurden 40 µl X-Gal-Lösung und 4 µl IPTG-Lösung pipettiert, mit einem sterilen Glas-'Spreader' ausplattiert und dem jeweiligen Transformationsansatz beimpft. Nach Inkubation bei 37°C für 12 bis 16 h wurde die Platte bis zur Entwicklung der Blaufärbung mehrere Stunden bei 4°C aufbewahrt. LacZ+-Kolonien waren durch eine blaue, lacZ--Kolonien durch eine weiße Färbung zu identifizieren.
Sowohl für die analytische, als auch für die präparative Gelelektrophorese wurden die Agarose-Gele zur Auftrennung von DNA-Fragmenten in 0.5´ TBE-Puffer zubereitet, der auch als Laufpuffer verwendet wurde. Die Konzentration der Agarose in den Gelen betrug je nach Molekulargewicht der aufzutrennenden DNA 0.6 bis 1.5 %. Nach Aufkochen der Gel-Lösung im Mikrowellenherd und kurzer Abkühlung auf 60°C wurde die flüssige Gelmasse direkt in eine Horizontalgelapparatur (11´15 cm bzw. 21´22 cm) bis zu einer Schichtdicke von 5-6 mm gegossen. War die Gellösung erkaltet und somit erstarrt, wurde das Gel dünn mit Laufpuffer überschichtet und der zur Bildung von Taschen eingesteckte Elektrophoresekamm herausgezogen. Vor dem Auftragen der DNA-Proben in die Geltaschen (1 bis 3 µg DNA) wurde die Probe mit 0.2 Vol. 6´ Gel-Ladepuffer vermischt. Neben den DNA-Proben wurden als Längenstandard die Kilobasenleiter bzw. HindIII geschnittene l-DNA aufgetragen (Tab.5; Abb.4).
Abbildung 4:l-HindIII (1)-u. KBL (2) -Standard
Tab. 5: Framentgrößen der verwendeten DNA-Längenstandards
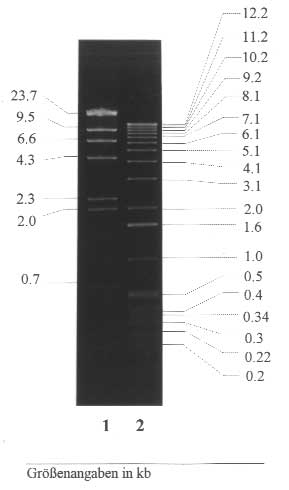 |
|
Die Elektrophorese wurde bei einer Gleichspannung von 5 V pro cm Elektrodenabstand durchgeführt. Die Laufstrecke der Farbmarker Bromphenolblau und Xylencyanol gaben einen Hinweis darauf, wann der Elektrophoreselauf abzubrechen war. Nach Auftrennung der Fragmente wurde das Gel 10 min in EtBr-Färbelösung gelegt. Das Bandenmuster wurde auf einem Transilluminator (HEROLAB UVT 2020) bei 254 nm sichtbar gemacht und mit einer POLAROID MP4 Kamera auf 665-POLAROID-Film dokumentiert. Die Fragmentgrößen wurden jeweils anhand der aufgeführten Längenstandards visuell bestimmt. In Einzelfällen wurde das Programm GELã zur Bestimmung der Fragmentgrößen verwendet.
3.8 Elution von DNA aus Agarosegelen
Zur Rückgewinnung von DNA aus Agarosegelen nach einer präparativen Gelelektrophorese wurde die 'Squeeze-Freeze' -Methode angewandt.
Die gewünschte Bande wurde unter UV-Licht mit einem Skalpell ausgeschnitten, wobei zu beachten war, daß die auszuschneidende Bande dem UV-Licht möglichst wenig ausgesetzt wurde, um eine UV-Schädigung der DNA zu vermeiden. Das ausgeschnittene Gelstück mit der gewünschten DNA-Bande wurde auf einer Glasplatte mit einer Rasierklinge zerkleinert und in ein Eppenporfgefäß gegeben. Dort wurden die Fragmente mittels eines Stößels weiter gequetscht und anschließend mit 100 µl TE-Puffer überschichtet. Nach Zugabe von 500 µl Phenol-Tris wurde der Ansatz dreimal in flüssigem Stickstoff schockgefroren und bei 37°C wieder aufgetaut. Zur Entfernung der Agarosetrümmer erfolgte eine 15minütige Zentrifugation bei 12 000 rpm und 4°C. Die DNA-haltige Oberphase wurde in einem neuen Eppendorfgefäß einer Phenolextraktion mit anschließender EtOH-Fällung unterzogen. In einem geeigneten Volumen resuspendiert, wurde 1 µl der DNA-Lösung auf einem Agarosegel überprüft.
3.9 Radioaktive DNA-Sequenzierung mit 35S
3.9.1 Sequenzierreaktionen
Die Sequenzierung von DNA wurde nach dem Kettenabbruch-Verfahren durchgeführt [Sanger et al., 1977].
Für die Sequenzierungsreaktion wurde der T7-Sequencing-Kit der Fa. LKB/Pharmacia und Plasmid-DNA, die nach den unter 3.1.2.1 und 3.1.3.1 beschriebenen Methoden isoliert wurde, verwendet. Zur Denaturierung der doppelsträngigen DNA wurden 2 µg Plasmid-DNA in einem Volumen von 8 µl mit 2 µl 2 N NaOH versetzt, gut gemischt, kurz abzentrifugiert und für 10 min bei RT inkubiert. Nach Volumenerhöhung durch Zugabe von 10 µl ddH2O wurde eine EtOH-Fällung durchgeführt. Von dem nun aufgereinigten Ansatz (12 µl) wurden 2 µl mittels Agarose-Gelelektrophorese überprüft. Nach dem positiven Ergebnis dieser Kontrolle schloß sich die 'Annealing-Reaktion' an. Dazu wurden zur DNA-Probe je 2 µl Primer und Annealing-Puffer gegeben und gut gemischt. Nach 20 min Inkubation bei 37°C und 10 min bei RT folgte die 'Labeling'-Reaktion.
Hierbei wurde der Ansatz mit 3 µl Labeling-Mix A (dCTP, dGTP und dTTP), 1 µl [a35S]dATPaS (entsprechend 10 µCi) sowie 2 µl (1.5 Units) T7-DNA-Polymerase versetzt. Nach fünfminütiger Inkubation bei RT wurden von diesem Reaktionsansatz je 4.5µl in bereits auf 37°C vorinkubierte Eppendorfgefäße aliquotiert, in die jeweils 2.5 µl des Sequenziergemisches mit den vier Desoxyribonukleosidtriphosphaten und einem der vier Didesoxynukleosidtriphosphate pipettiert worden waren. Die vier Sequenzierreaktionen wurden 5 min bei 37°C im Wasserbad inkubiert. Um die Reaktion abzustoppen, wurden jedem Reaktionsansatz 5 µl Stop-Lösung zugesetzt. Bis zum späteren Gebrauch wurden die Ansätze bei -80°C aufbewahrt.
3.9.2 Polyacrylamid-Gelelektrophorese
Die Auftrennung der Proben erfolgte in 6 % Polyacrylamid-Harnstoffgelen unter Verwen-dung einer Elektrophoreseapparatur der Fa. LKB/Pharmacia.
Vor dem Gießen des Sequenziergels wurden die Thermostatisierplatte und die Gel-Trägerplatte mit einem Detergenz gründlich gereinigt und mit EtOH nachbehandelt. Um das spätere Ablösen des Gels von der Thermostatisierplatte zu erleichtern, wurde diese mit 5 ml Repel-Silan behandelt. Auf die Gelträgerplatte wurden 5 ml Bindesilan aufgetragen, um die Haftung des Gels an der Glasplatte zu gewährleisten. Zur Herstellung der Gel-Lösung wurden ca. 30 ml Polyacrylamidlösung mit 30 µl TEMED und 300 µl Ammoniumpersulfat versetzt und unter Verwendung eines Taschenkammes wurde diese Gel-Lösung mit einer Schichtdicke von 0.2 mm zwischen die vorbereiteten Glasplatten gegossen. Nach einer Polymerisationszeit von mindestens 2 h wurde das Gel in die Apparatur eingesetzt, die Kammern mit TBE-Puffer gefüllt und die Thermostatisierplatte auf 60°C beheizt. Nach Ziehen des Kammes wurden die Geltaschen mit TBE-Puffer gespült und der Vorlauf über 60 min bei 30 V/cm gestartet. Danach wurden die Geltaschen erneut mit TBE-Puffer gespült und 1.5 µl der Proben, die zuvor 3 min bei 95°C denaturiert und auf Eis abgekühlt worden waren, mittels einer Hamiltonspritze aufgetragen. Der anschließende Lauf des Sequenziergels erfolgte bei 60°C und 40 V/cm. Um mehr Sequenzinformation zu erhalten wurde das Auftragen der Proben 2-3 mal im Abstand von 3-4 h, je nach der Laufstrecke der Bromphenol- und Xylencyanol-Farbmarker, wiederholt.
Nach Ende der Elektrophorese wurde die Gel-Trägerplatte mit dem anhaftenden Polyacrylamidgel von der Thermostatisierplatte abgehoben, zur Entfernung des Harnstoffes 30 min in 10 % (v/v) Essigsäure getaucht, gründlich mit demineralisiertem Wasser gespült und anschließend mindestens 1 h bei 65°C getrocknet.
3.9.3 Autoradiographie
Im Anschluß an die Polyacrylamid-Harnstoff-Gelelektrophorese wurde zur Autoradio-graphie ein Medical-X-Ray Screenfilm der Fa. CeaAB verwendet.
Die Autoradiographie erfolgte durch Auflegen des Films auf das getrocknete Sequenzgel in einer lichtundurchlässigen Exponierbox über einen Zeitraum von 2-6 Tagen. Der exponierte Film wurde anschließend 5 min entwickelt (KODAK LX-24-Entwickler), 1 min in 3 % (v/v) Essigsäure geschwenkt, 5 min fixiert (KODAK LX-24-Fixierer), gründlich mit demineralisiertem Wasser abgespült und getrocknet.
3.9.4 Analyse der Sequenzdaten
Nach Einlesen der Sequenzdaten erfolgte die Auswertung mit Hilfe des GCG-Programmpaketes der University of Wisconsin Genetics Computer Group [Devereux et al., 1984]. Neben einer großen Anzahl von GCG-Programmen wurden zusätzlich dem GCG-Paket nachträglich assoziierte Programme verwendet; in diesem Zusammenhang sei das Programm FOLD [Zuker, 1981] erwähnt, mit dem RNA-Sekundärstrukturen entwickelt wurden. Datenbankrecherchen wurden mit dem Programmpaket HUSAR2.0 (Heidelberg Unix Sequence Analysis Resources) des Deutschen Krebsforschungszentrums Heidelberg und des Molekularbiologischen Zentrums/Universität Heidelberg durchgeführt.
Zur Entwicklung phylogenetischer Beziehungen, basierend auf multiplen Alignments, wurde das Programmpaket TREE, enthalten im HUSAR2.0-Programmpaket, verwendet [Feng u. Doolittle, 1987].
Zum Nachweis von Helix-Turn-Helix-Motiven in einem offenen Leserahmen wurde die von Dodd u. Egan beschriebene Methode angewandt [Dodd u. Egan, 1987]. Alle Proteine, für die bisher spezifische DNA-Bindungseigenschaften nachgewiesen wurden, zeigen ein sogenanntes Helix-Turn-Helix-Motiv. Wie Röntgenstrukturanalysen ergeben haben, handelt es sich dabei um eine Sekundärstruktur der Peptidsequenz, die die sterische Bindung des Proteins an den DNA-Doppelstrang ermöglicht [Pabo u. Sauer, 1984]. Zur Auffindung des Helix-Turn-Helix-Motivs ausgehend von einer DNA-Sequenz wurde die entsprechende Basenfolge mit Hilfe des GCG-Programms TRANSLATE in die entsprechende AS-Folge übersetzt. Nach einem Alignment mit homologen Peptidsequenzen, für die bereits Helix-Turn-Helix-Motive postuliert waren, konnte der Bereich eines solchen Motivs in der zu untersuchenden Sequenz eingegrenzt werden. Der spezifische Nachweis des Motivs erfolgte nach der Methode von Dodd u. Egan für l-Cro-ähnliche DNA-Bindungsregionen in Proteinen unter Verwendung der angegeben Tabellen zur Frequency- und Weight-Matrix, sowie zur Score-Wertung [Dodd u. Egan, 1987]. Nach Vergleich der hochkonservierten AS der Frequency-Matrix mit der im einzelnen zu untersuchenden Peptidesquenz wurde für jede AS in diesem Segment der entsprechende Weight-Matrix-Wert abgelesen. Die Summe aller Werte der einzelnen AS ergibt das Score-Ergebnis - ein relatives Maß für die Wahrscheinlichkeit, daß tatsächlich eine Helix-Turn-Helix-Struktur vorliegt.
3.10 Nachweis von homologen DNA-Sequenzen
3.10.1 Techniken zum DNA-Blotting
3.10.1.1 Southern-Blot
Zur Lokalisierung spezifischer DNA-Sequenzen wurde die Methode nach Southern mit einigen Modifikationen verwendet [Southern, 1975].
Nach elektrophoretischer Auftrennung von DNA-Fragmenten im Agarose-Gel, Anfärben in EtBr-Färbelösung und photografischer Dokumentation wurde die DNA durch Southern-Transfer mittels einer Vakuum-Blot-Anlage (VacuGene™ X1, LKB/Pharmacia) auf eine Nylonmembran (PALL BioSupport™) übertragen. Zu diesem Zweck wurde der Nylonfilter auf eine poröse Trägerplatte gelegt, der Rand um den Filter mit einer Folienmaske abgedeckt und das Gel auf dem Filter plaziert. Durch Schließen der Klammern am Rahmen wurde die Apparatur abgedichtet und nach Anlegen eines Vakuums von 40-50 mbar wurden nacheinander für je 20 min 30 ml 0.25 N HCl, Denaturierungs- und Neutralisierungslösung aufgetragen. Der eigentliche Transfer erfolgte durch Überschichtung des Gels mit 20´ SSC über einen Zeitraum von 1 h. Nach Beendigung des Transfers wurde die Nylonmembran 5min in 2´ SSC gewaschen und 10 min bei 40°C im Vakuumofen getrocknet. Die kovalente Bindung der DNA an die Filtermatrix erfolgte mittels UV-'Cross-Linking', wobei die Membran für 3 min auf dem UV-Transilluminator bestrahlt wurde.
3.10.1.2 Dot-Blot
DNA-Proben wurden wie unter 3.9.1 beschrieben denaturiert und nach EtOH-Fällung in einem Volumen von 2 µl ddH2O aufgenommen. Von dieser DNA-Lösung wurde 1 µl auf eine trockene Nylonmembran (PALL BioSupport™) getropft und nach zehnminütiger Trocknung bei 40°C im Vakuumofen erfolgte die Fixierung der DNA mittels UV-'Cross-Linking' (3 min UV-Transilluminator). Für das Blotting größerer Volumina wurde die DNA-Probe in einer Hybrislot-Apparatur mittels Unterdruck auf den Filter gesaugt.
3.10.2 Herstellen einer DNA-Sonde
3.10.2.1 Markierung mit Digoxigenin-11-dUTP
Die Markierung der DNA-Sonden, sowie Sondenkontrolle, Hybridisierung und Entwicklung des Filters wurde mit dem DNA Labeling and Detection Kit nonradioactive der Fa. BOEHRINGER/Mannheim durchgeführt. Dabei ermöglicht die Markierung einzelsträngiger DNA mit Digoxigenin im 'Random-Priming' -Verfahren die spätere Anfärbung und Lokali-sierung dieser markierten Sonde auf dem Hybridisierungsfilter und somit die Lokalisierung homologer DNA-Bereiche.
Zur Markierung der Sonde wurden 10 ng bis 3 µg doppelsträngige DNA in einem Volumen von 15 µl 10 min im kochenden Wasserbad denaturiert und im Eisbad abgekühlt. Zu der nun einzelsträngigen DNA wurden 2 µl Hexanukleotidgemisch ('Random-Priming'), 2 µl dNTP Labeling-Gemisch (enthält neben den vier Desoxynukleotiden Digoxigenin-11-dUTP)und 1 µl (2 Units) Klenow-Enzym pipettiert. Der Ansatz wurde kurz abzentrifugiert und mind. 60 min bei 37°C inkubiert. Durch Zugabe von 2 µl 0.2 M EDTA-Lösung wurde die Reaktion abgestoppt und nach dem Zusatz von 2.5 µl 4 M LiCl-Lösung schloß sich eine EtOH-Fällung an, wobei jedoch kein Natriumacetat zugegeben wurde. Das erhaltene DNA-Pellet wurde in 50 µl TE-Puffer resuspendiert und die nun fertige Digoxigenin-markierte Sonde bei -20°C gelagert.
3.10.2.2 Kontrolle der Sonde
Zur Bestimmung der Sondenkonzentration wurde eine Verdünnungsreihe der Dig-markierten Test-DNA des Herstellers, sowie Digoxigenin-markierte Sonden-DNA mittels Dot-Blot auf eine Nylonmembran übertragen. Hierzu wurden von jeder DNA-Lösung jeweils 1 µl auf die Membran pipettiert und nach der vom Hersteller empfohlenen Methode angefärbt. Der Vergleich der Farbintensität von Sonde mit den Verdünnungsstufen ermöglichte die Bestimmung der Konzentration an markierter Sonden-DNA.
3.10.3 DNA-DNA-Hybridisierung
Die Hybridisierung der unter 3.10.2 Digoxigenin-markierten Sonde mit einer denaturierten DNA-Probe, die auf einer Nylonmembran fixiert wurde und auf zur Sonde homologe DNA-Bereiche untersucht werden sollte, ermöglichte die spätere Lokalisierung eben dieser Homologiebereiche mittels spezifischer Farbreaktionen. Die im folgenden angegebenen Mengenangaben beziehen sich jeweils auf 100 cm² Filterfläche. Die Hybridisierungs- und Waschschritte wurden in einer SCHOTT-Hybridisierungsröhre in einem Hybridisierungs-ofen der Fa. BACHOFER durchgeführt.
Zur Absättigung unspezifischer Bindungen wurde die Nylonmembran mit den denaturierten DNA-Proben 1 h bei 42°C mit 20 ml Prähybridisierungslösung inkubiert. Anschließend wurde diese Lösung gegen die Hybridisierungslösung, die 2.5 ml Prähybridisierungslösung und 50 µl markierte und frisch denaturierte (10 min bei 95°C) Sonden-DNA enthielt, ersetzt. Der Filter wurde mit der Hybridisierungslösung über Nacht bei 42°C inkubiert, 2 ´ 5 min bei RT mit Wasch-Lösung I und 2 ´ 15 min bei 68°C mit Wasch-Lösung II gespült. Die Hybridisierungslösung wurde bei -20°C gelagert und mehrfach wiederverwendet.
3.10.4 Detektion
Der Nachweis der durch Hybridisierung gebundenen, markierten Sonden-DNA erfolgte durch eine enzymatische Farbreaktion. Zunächst wird der Filter mit alkalischer Phosphatase, die mit einem Digoxigenin-Antikörper verknüpft ist, inkubiert. Der Antikörper bindet spezifisch an das Digoxigenin der Sonde und die über den Antikörper gebundene alkalische Phosphatase oxidiert die anschließend zugegebene farblose Verbindung X-Phosphat (5-Brom-4-chlor-3-indolylphosphat) durch Abspaltung der Phosphorylgruppe zu blaugefärbtem Indigo. In einer enzymatisch gekoppelten Reaktion wird NBT (Nitroblau-tetrazolium-chlorid) zu Diformazan reduziert. Beide Verbindungen führen zur Bildung eines blauen, unlöslichen Farbniederschlages auf dem Nylonfilter.
Die folgenden Schritte zur Farbentwicklung auf dem Filter wurden bei RT durchgeführt. Zu Beginn der Entwicklung wurde die Nylonmembran 1 min mit Detect-Puffer I gewaschen, 30 min in Detect-Puffer II inkubiert und erneut kurz mit Detect-Puffer I gespült. Nach 1 h Inkubation mit Antikörper-Lösung wurde der Filter 2 ´ 15 min mit Detect-Puffer I gewaschen, zur Einstellung eines alkalischen pH-Wertes 2 min mit Detect-Puffer III gespült und luftblasenfrei mit 5 ml Färbelösung in einer Kunststoff-Folie eingeschweißt. Nach Auftreten der Signalbanden wurde die Farbreaktion durch Spülen der Membran in TE-Puffer oder demineralisiertem Wasser abgestoppt.
Das Ergebnis wurde mit einer POLAROID-Sofortbildkamera und Positiv/ Negativfilm 665 bei Blende 16 und 1 sec Belichtungszeit photographisch dokumentiert.
<< nach oben
zurück